Date: April, 2024 | Category: Compliance | Author: Hana Trokic
Understanding the complex regulatory compliance landscape of the European Union’s (EU) pharmaceutical industry poses a significant challenge for companies aiming to remain compliant while ensuring the highest standards of product quality and safety.
The stringent regulatory framework for EU pharma, designed to safeguard public health, mandates adherence to rigorous regulatory compliance with the European Medicines Agency (EMA), Heads of Medicines Agencies (HMAs), the European Commission, and guidelines such as the Good Manufacturing Practice (GMP), and the Good Clinical Practice (GCP), among other requirements.
By exploring the pivotal role of technology in enhancing compliance, and highlighting the importance of quality control systems like automated proofreading technology, this guide serves as a roadmap for pharmaceutical companies striving to navigate the demanding regulatory environment of the EU.
Understanding EU Pharmaceutical Regulatory Compliance
The foundation of staying compliant begins with a thorough understanding of the relevant regulations in the European Union.
The EU has established a comprehensive regulatory compliance framework that includes directives and regulations from the above-mentioned health authorities, along with specific national regulations of member states.
All of these guidelines are in place to ensure the safety, efficacy, and quality of pharmaceutical products, subsequently ensuring their proper use and the well-being of consumers and patients. These guidelines cover a broad spectrum, from clinical trials to marketing authorizations, pharmacovigilance, and manufacturing practices.
Key EU Pharmaceutical Guidelines
Key pharmaceutical guidelines for the EU are enforced by the European Medicines Agency (EMA) and national regulatory agencies within member states, aiming to harmonize regulatory compliance standards across the EU and ensure the highest level of protection for public health.
Some key guidelines include:
- Good Manufacturing Practice (GMP): Any manufacturer of medicine intended for the EU market, regardless of its global location, must adhere to GMP standards, ensuring that the medicines are of consistently high quality, appropriate for their intended use, and meet the requirements of the marketing authorisation or clinical trial authorisation.
- Good Clinical Practice (GCP): Represents an ethical and scientific quality benchmark for the design, recording, and reporting of trials involving human subjects, ensuring public confidence by protecting the rights, safety, and wellbeing of participants and guaranteeing the credibility of clinical trial data.
- Good Laboratory Practice (GLP): Defines a set of rules and criteria for a quality system focused on the organizational process and conditions under which non-clinical health and environmental safety studies are systematically planned, executed, monitored, documented, reported, and archived.
- Good Distribution Practice (GDP): Ensures that medicines are legally authorized, correctly stored and transported, free from contamination, properly rotated in storage, and delivered on time to the right recipient. It also mandates a tracing system for identifying and recalling faulty products. Additionally, GDP covers the proper handling of active ingredients and other materials used in making medicines.
- Clinical Trials Regulation (EU) 536/2014: Aims to unify clinical trial rules across the EU by introducing a single submission and authorization process through an EU portal, a uniform assessment leading to one decision, and clear rules on participant protection, informed consent, and transparency.
- Pharmacovigilance Legislation: Enacted in July 2012, this legislation mandates reporting and monitoring of adverse effects from medical products to ensure patient safety. It also establishes regulatory frameworks for assessing risks, managing safety information, and communicating it to healthcare professionals and the public.
- Advanced Therapy Medicinal Products (ATMPs): ATMPs are divided into three main categories:
- Gene therapy medicines: includes genes that cause therapeutic, preventive, or diagnostic effects by introducing lab-created DNA into the body to treat diseases like genetic disorders, cancer, or chronic illnesses.
- Somatic-cell therapy medicines: consist of cells or tissues that have been altered in their biological characteristics or are used in different ways from their original functions in the body, aiming to treat, diagnose, or prevent diseases.
- Tissue-engineered medicines: involve cells or tissues modified to repair, regenerate, or replace human tissue.
Leveraging Technology for Regulatory Compliance
Having gone through key pharmaceutical regulations in the European Union, let’s explore how technology helps organizations adhere to them to ensure the regulatory compliance of their products. To do this, technology plays a critical role in achieving and maintaining compliance, especially in regulated industries such as pharmaceuticals.
Automated systems and advanced technologies such as Artificial Intelligence (AI) and Machine Learning (ML) are being leveraged to streamline regulatory compliance processes throughout the pharmaceutical and drug development lifecycle. These algorithms analyze vast amounts of data to identify patterns, deviations, and potential risks in manufacturing processes, enabling proactive intervention to prevent compliance breaches.
For perspective, GlobalVision’s independent market research has recently shown that AI-powered technologies help pharmaceutical companies get their products to market faster. Our survey results revealed that 89% of regulatory survey respondents said that using AI-powered solutions significantly reduced their compliance review process, allowing them to deliver products to market faster.
This goes hand in hand with recent plans being put into place with regards to the use of AI in EU pharma. As it is predicted to be so substantial in the near future, the EMA and the HMA have published an artificial intelligence work plan to 2028, setting out a collaborative and coordinated strategy to maximize the benefits of AI to stakeholders while managing the risks.
Along with AI technologies, other solutions to consider are quality control systems that help organizations stay compliant by easing the regulatory compliance process, and improving processes, and also cloud-based solutions that enable data sharing among stakeholders to maintain data integrity and confidentiality.
It is also important to take into consideration solutions that have gone through a validation process. Validation involves a series of activities that take place over the lifecycle of any product and process to ensure that it has the required quality and is satisfying all requirements before being used.
By integrating these technologies into regulatory compliance frameworks, and taking into consideration certain features, EU pharmaceutical companies can enhance regulatory compliance, minimize risks, and ultimately improve the quality of their products.
The Right Solution for Regulatory Compliance – Automated Proofreading Software
Choosing the right technological solution is essential for ensuring compliance with EU regulations. But one that incorporates many solutions and leverages many technologies may be the best way to go. This is where automated proofreading software comes into play.
Automated proofreading is an innovative quality control solution for pharmaceuticals that helps maintain the accuracy and quality of documents and subsequent products by inspecting all types of regulatory content present in pharmaceuticals.
These documents include, packaging inserts and leaflets, product labels and packaging, regulatory submissions, clinical trial protocols and reports, Standard Operating Procedures (SOPs), and marketing materials such as brochures, websites, and promotional videos.
Regardless of the type of file or document, the advanced system helps pharmaceutical companies in the EU uphold quality standards with greater ease and efficiency, ensuring regulatory compliance within the EU.
This technological innovation provides better proofreading results and empowers teams to produce compliant and error-free documents as it acts as an added layer of insurance in quality control processes. Some benefits of automated proofreading include:
- Proofreads critical content at scale: Automates proofreading of large volumes of documents at lightning speed, saves valuable time and resources and accelerates company growth while maintaining quality assurance.
- Mitigates risk of financial losses: Automatically identifies critical errors such as incorrect drug names, dosages, and safety warnings and minimizes the risk of costly errors like product recalls, legal disputes, regulatory fines, reputational damage.
- Expedite time-to-market: Increases speed of review and approval cycles for regulatory submissions, labeling, packaging, marketing materials and more and allows organizations to gain a competitive market advantage.
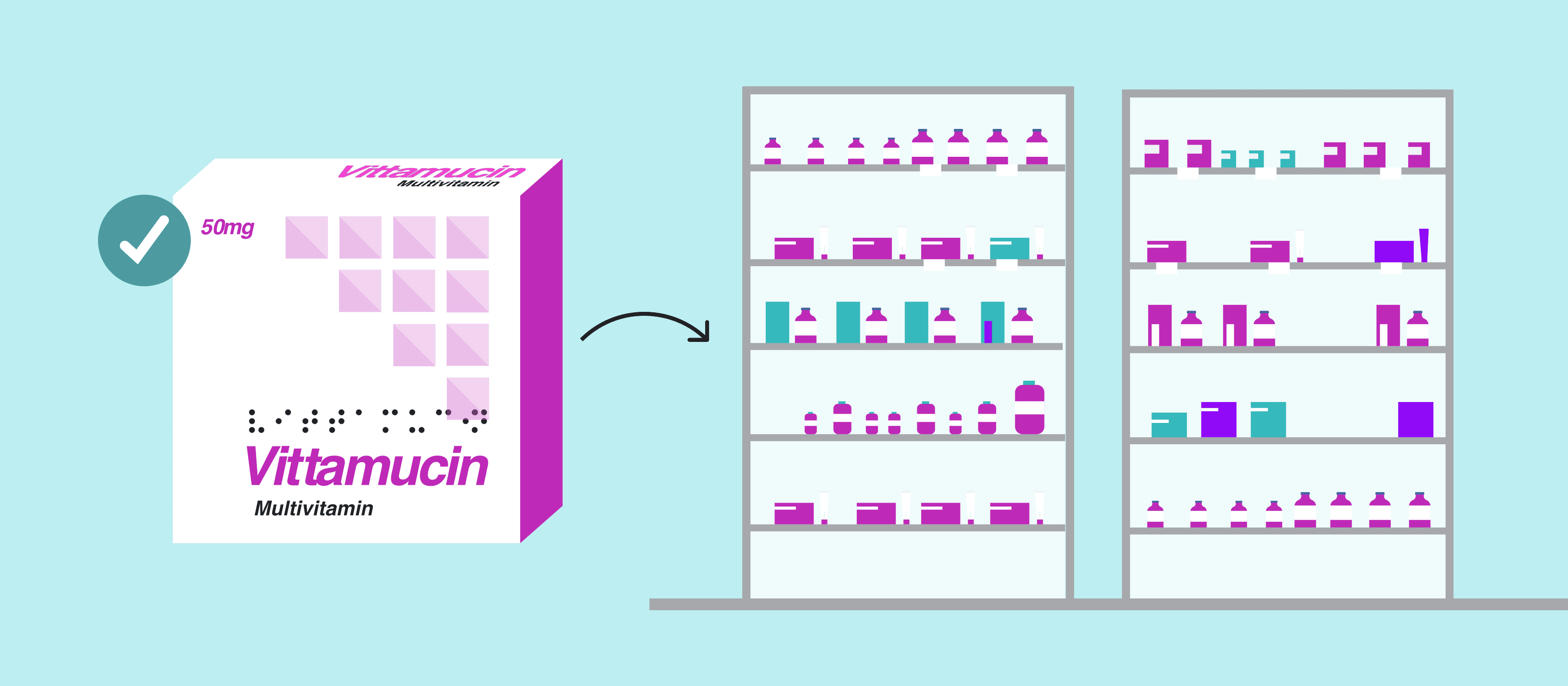
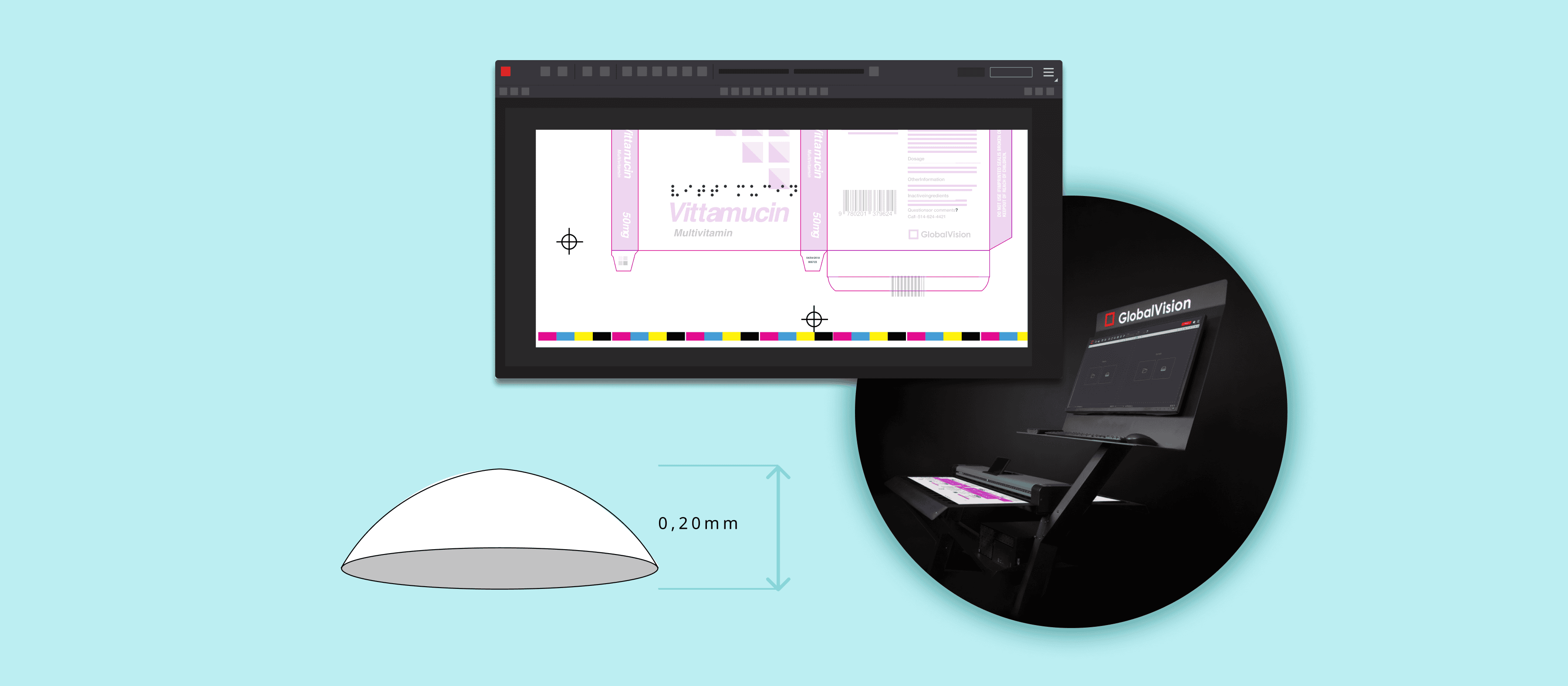
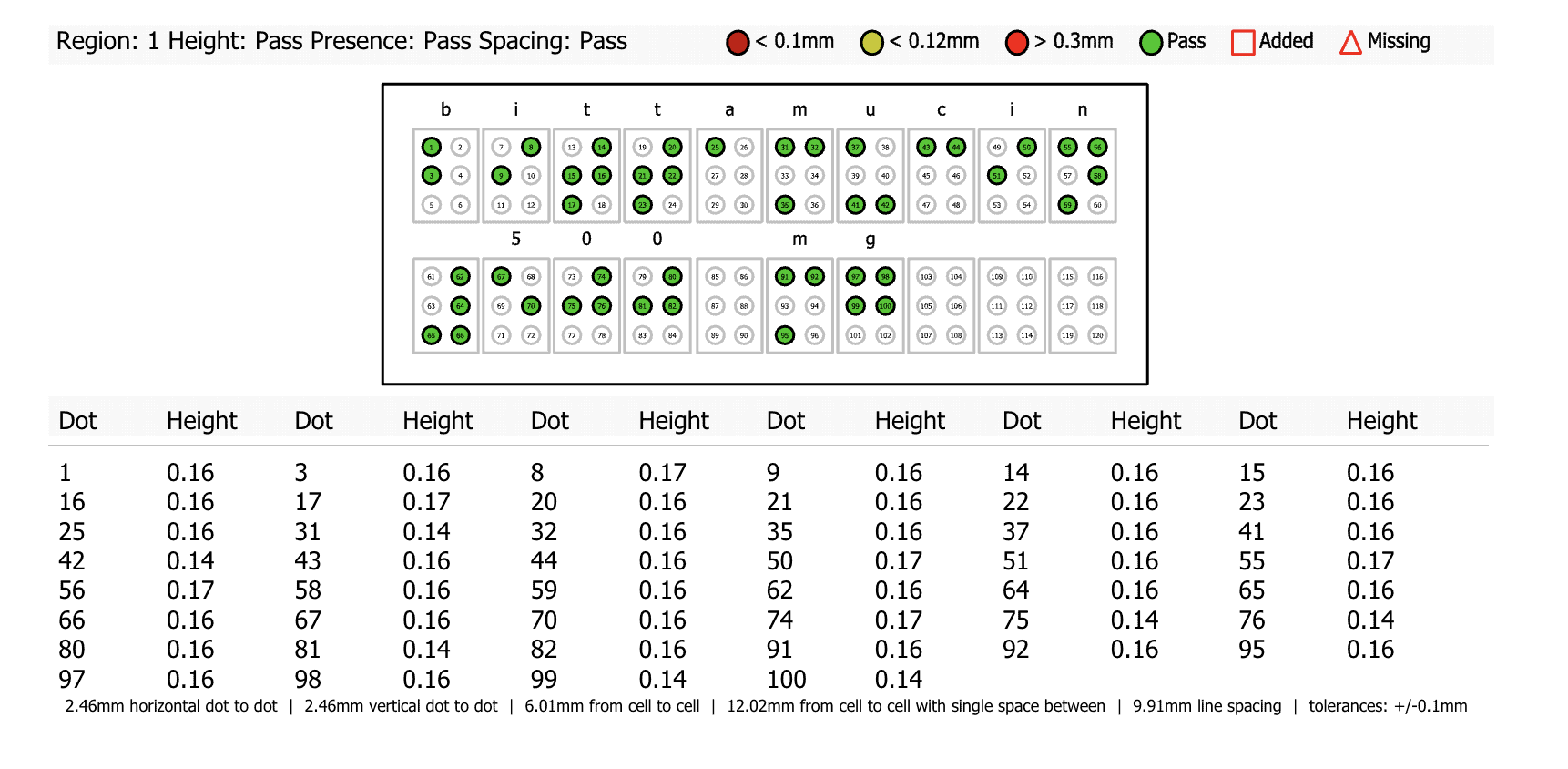
In essence, automated proofreading works by comprehensively inspecting documents to find discrepancies in the file. The innovative software conducts detailed inspections and proofreads all packaging assets from text, color, graphics, barcodes, braille, and more.
Through automated proofreading, inspections of packaging components are completed exponentially faster than traditional proofreading, and a task that once took hours or days to complete is now reduced to only a few minutes.
“The biggest benefit [of GlobalVision] to our facility is the reduction in the amount of processing and testing time (by at least a factor of 3) and the ability to receive competent and timely support when needed….Overall, the greatest benefit is being able to rely on a high-quality product that allows us to be more efficient.”
- Veronica Guilliams, Senior Manager – Visual Inspection, Pfizer
Why Automated Proofreading is Essential for EU Regulatory Compliance
Automated proofreading software plays a crucial role in enhancing the quality and regulatory compliance of pharmaceutical products in the EU. Some reasons why automated proofreading is an essential tool and software to leverage are:
- Error Detection: Swiftly identify potential errors on pharmaceutical files, guaranteeing both accuracy and quality. This is vital not only for regulatory adherence but also for safeguarding patient safety. Accurate content ensures vital product information such as ingredients, dosages, and expiry dates are correctly portrayed, mitigating potential risks and upholding user well-being.
- Consistency: Verify consistency in terminology, dosage information, and instructions throughout pharmaceutical documentation. Consistent information reduces confusion among healthcare professionals and users, enhancing clarity and usability.
- Regulatory Compliance: Adhering to stringent regulatory requirements is critical in the pharmaceutical industry. Automated proofreading ensures all content aligns with regulatory standards, minimizing the risk of non-compliance issues such as fines, recalls, or legal repercussions.
- Efficiency and Speed: Streamlines the proofreading process compared to manual reviews and methods, offering significant efficiency gains. This speed offers a particular advantage in the dynamic and fast-paced pharmaceutical industry, where timely and accurate product releases are crucial to get oftentimes life-saving pharmaceuticals to market and to capture significant market share.
- Version Control: Facilitate effective version control, ensuring that all documents reflect the most current and accurate information. This mitigates risks associated with outdated content, maintaining precision and relevance.
- Consistent Branding: For pharmaceutical companies with diverse product portfolios, maintaining consistent branding across all products and content is essential for brand recognition and trust. Automated proofreading aids in upholding this consistency, reinforcing brand integrity.
By leveraging automated proofreading, EU pharmaceutical companies can elevate the accuracy, quality, and regulatory compliance of their products. This ultimately not only enhances patient safety but also preserves the integrity of medical products, aligning with corporations commitment to excellence and safety in healthcare.
GlobalVision’s Verify For Complete EU Regulatory Compliance
Verify, is GlobalVision’s cloud-based proofreading platform that is tailored specifically for the needs of regulated industries in the European Union such as pharma.
The software has been meticulously evaluated and its functionality, performance, security, and compliance with relevant standards and regulations in the EU is guaranteed as the software fits the needs of end-users and helps adhere to industry-specific regulatory compliance requirements.
As a solution created for regulatory professionals, teams, and corporations, Verify seamlessly integrates with existing workflows and acts as an added layer of insurance that mitigates the risk of errors slipping through in highly-regulated documentation.
The software allows for comprehensive inspections with robust capabilities including text and graphics comparisons, spell check, braille, and barcode inspections, all critical components of EU pharmaceutical documentation. What’s more, these comprehensive inspections are done in record-time and eliminate the need for tedious, manual reviews.
This speed and precision in document inspections allow for faster review and approval cycles, enabling EU-compliant products to reach the market quickly, subsequently capturing market share from competitors due to their expedited workflows and allowing for a competitive advantage.
Conclusion
For the pharmaceutical industry, navigating the complex regulatory landscape of the European Union is no easy task and demands commitment to compliance.
The strict regulations set forth by governing health authorities in the EU are in place to help ensure the safety and efficacy of medicinal products while subsequently ensuring the safety of the end user.
Once a tedious and time-consuming task, adhering to regulations becomes easier when corporations turn to technology to help them adhere to regulatory compliance
Leveraging technology not only increases the speed at which quality control processes can be undergone, but also increases the accuracy and efficiency. Automated proofreading in particular, stands out as an ideal technological solution for EU pharmaceutical companies, offering advantages to document reviews such as error detection, speed, consistency, and version control, to name a few.
By integrating this technology, companies can not only meet EU regulatory compliance requirements but also uphold the highest standards of product quality and safety, thereby reinforcing their commitment to excellence in healthcare and ensuring the well-being of consumers and patients alike.
If you want to begin ensuring the regulatory compliance of your EU pharma documentation, book a demo of Verify and see firsthand the transformative impact it will have on your pharmaceutical proofreading and quality control processes.
Also, read our Pharmaceutical Industry Report to get exclusive insights into the growing role of technology in regulatory affairs and see how other top industry leaders are leveraging this technology to help them achieve their business goals.